


| 图片 | 品牌 | 名称 | 货号 | 规格 | 赛恩斯价 | 库存 | 货期 | 购买 |
|---|
Gaussian 16 是高斯系列电子结构程序的最新版本,被世界各地的化学家、化学工程师、生物化学家、物理学家和其他科学家使用。Gaussian 16 提供了一套广泛的最先进的建模功能。您可以使用它来研究您感兴趣的现实世界中的化学问题,这些问题非常复杂,即使在普通的计算机硬件上也是如此。
Gaussian 16 与其他软件有何不同?
基本功能
高斯 16 从量子力学的基本定律出发,预测了化合物和反应在各种化学环境中的能量、分子结构、振动频率和分子性质。Gaussian 16 模型既可以应用于稳定物质,也可以应用于难以或不可能通过实验观察到的化合物,无论是由于它们的性质(例如,毒性、可燃性、放射性)还是它们固有的短暂性质(例如,短寿命中间体和过渡结构)。
使用 Gaussian 16,您可以彻底研究您感兴趣的化学问题。例如,您不仅可以快速可靠地最小化分子结构,还可以预测过渡态的结构,并验证预测的稳定点实际上是最小值或过渡结构(视情况而定)。您可以继续按照本征反应坐标 (IRC) 计算反应路径,并确定哪些反应物和产物通过给定的过渡结构连接。一旦你对势能表面有了完整的了解,就可以准确预测反应能和势垒。您还可以预测各种各样的化学性质。
Gaussian 16 提供了多种用于模拟化合物和化学过程的方法,包括:
Gaussian 16 中的分子性质
反铁磁耦合
原子电荷
溶剂化
ΔG 偶极矩
电子亲和力
电子密度
电子圆二色性 (ECD)
静电势
静电势衍生电荷
电子过渡带形状
高精度能量
超精细耦合常数(各向异性)
超精细光谱张量(包括g 张量)
电离电位
红外和拉曼光谱*
共振前拉曼光谱*
共振拉曼光谱
分子轨道
多极矩
NMR 屏蔽和化学位移
NMR 自旋-自旋耦合常数
旋光度 (ORD)
极化率/超极化率
拉曼光学活性 (ROA)*
热化学分析
紫外/可见光谱
振动-旋转耦合
振动圆二色性 (VCD)*
振动(吸收和发射)光谱
*谐波近似值,包括非谐波效果
E能量;G解析梯度;F解析频率;F†使用解析频率重新实现。
使用 GaussView 的可视化功能可以检查各种高斯结果:
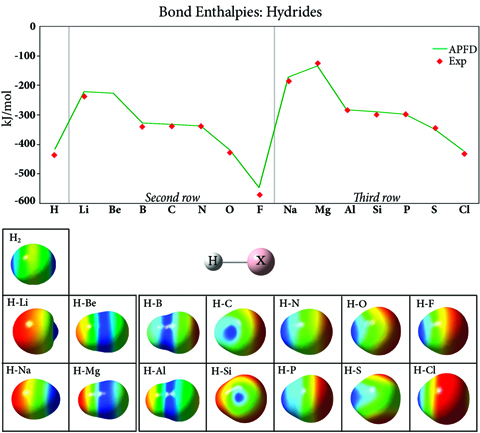
氢化物中的键焓和静电势
该图绘制了第二行和第三行氢化物化合物的键强度(实验:[CRC00]),该强度通常在整个元素周期表中增加,最强的键出现在惰性气体之前的元素中。该图的两行具有相似的整体形状,但由于填充的第二个壳对原子核的额外屏蔽,第三行的值更高。这些图像显示了映射到等密度表面上的每种化合物的静电势。The H2表面说明了这种键的共价性质;其他氢化物化合物中的键是离子的。负静电势(红色)位于每行开头的氢原子上,并且随着一行内原子序数的增加,它移动到取代基。因此,由于电负性的变化,氢化物键强度在一个周期(行)上增加,随着你向下移动一组(列)而降低。
| 象形图 | |
|---|---|
| 警示语 | |
| Class | |
| 警告声明 | |
| UNub | |
| 危险声明 | |
| Packing Group |

 扫码关注
扫码关注

